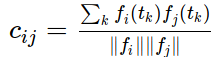R package Tutorial
Introduction
The R package tutorial can ve directly viewed in the R console with the command browseVignettes("cliqueMS") assuming you have
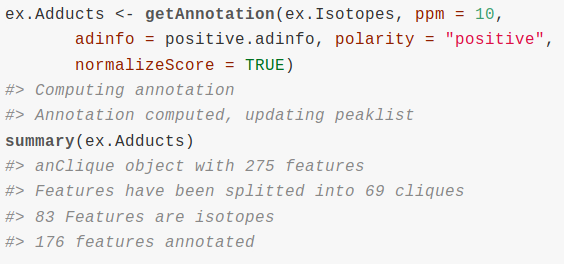
Untargeted metabolomics goal is to quantify as metabolites as possible from a sample. We can use liquid chromatography coupled to mass spectrometry (LC/MS) for this purpose. It is a great challenge to transform LC/MS data into a profile of annotated metabolites that provides us meaningful biological information. A very important limitation to the annotation of metabolomic experiments is that the number of m/z processed signals, called features, is much bigger than the putative number of metabolites in a sample. The sources that produce multiple features from a single metabolite are multiple and variable. Natural isotopes as carbon isotopes produce isotope features. Ionization of metabolites produce the so called adducts of the metabolite, which are detected as different features depending on the ion adduct involved ([M+Na]+, [M+H]+, etc..). Apart from adduct features, ionization also produces metabolite fragmentation and other reactions as dimerizations, trimerizations, all of them being detected as multiple features. Being the reduction of multiple features to single metabolites a crucial step for the correct annotation of LC/MS experiments, we will show how to use cliqueMS to do so.